6.3 相关性分析 EMP_cor_analysis
多组学数据分析中,经常通过相关系数矩阵来观察特征之间的相互关系。
本模块在计算相关性时,会自动选择两个组学项目数据中无缺失值的交集进行分析。
6.3.1 探索微生物数据与样本相关数据的相关性
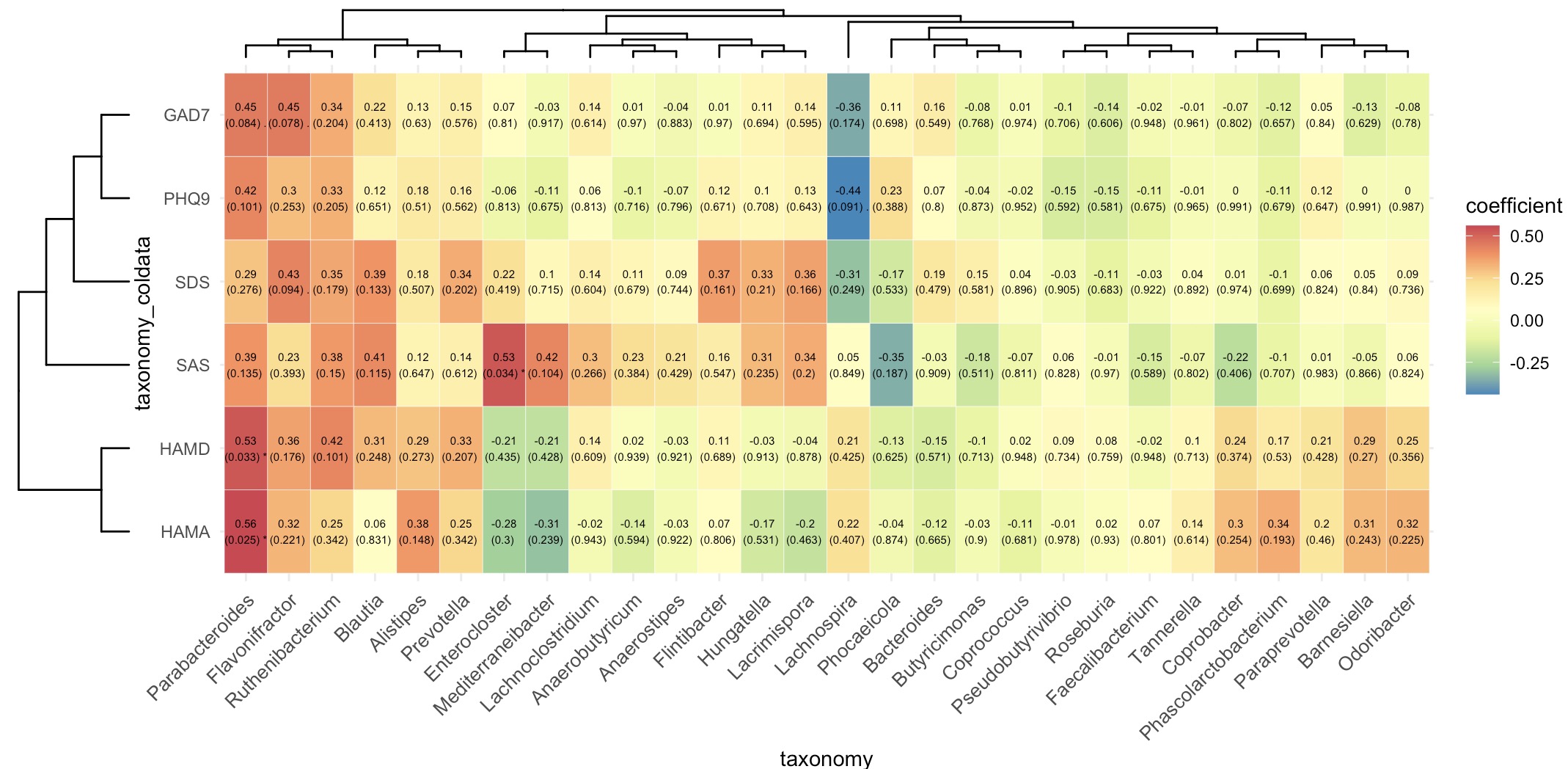
🏷️示例1:微生物物种注释数据与量表评分数据的相关性分析。
提取组学项目taxnomy的assay,利用模块EMP_identify_assay筛选核心微生物,利用模块EMP_collapse折叠出属级别数据,利用模块EMP_decostand进行相对丰度的标准化。
micro_data <- MAE |>
EMP_assay_extract('taxonomy') |>
EMP_identify_assay(method='default') |>
EMP_collapse(estimate_group = 'Genus',collapse_by = 'row') |>
EMP_decostand(method='relative')
micro_data

然后,从MAE对象中提取组学项目taxnomy的assay,并进一步提取对应的coldata的量表评分数据。
meta_data <- MAE |>
EMP_assay_extract('taxonomy') |>
EMP_coldata_extract(action = 'add',
coldata_to_assay = c('SAS','SDS','HAMA','HAMD','PHQ9','GAD7'))
meta_data

最后,使用+符号将微生物丰度数据和量表评分数据合并成EMP对象,使用EMP_cor_analysis和EMP_heatmap_plot模块完成相关性分析及可视化。
(micro_data + meta_data) |>
EMP_cor_analysis() |>
EMP_heatmap_plot(label_size=2,palette='Spectral',
clust_row=TRUE,clust_col=TRUE)
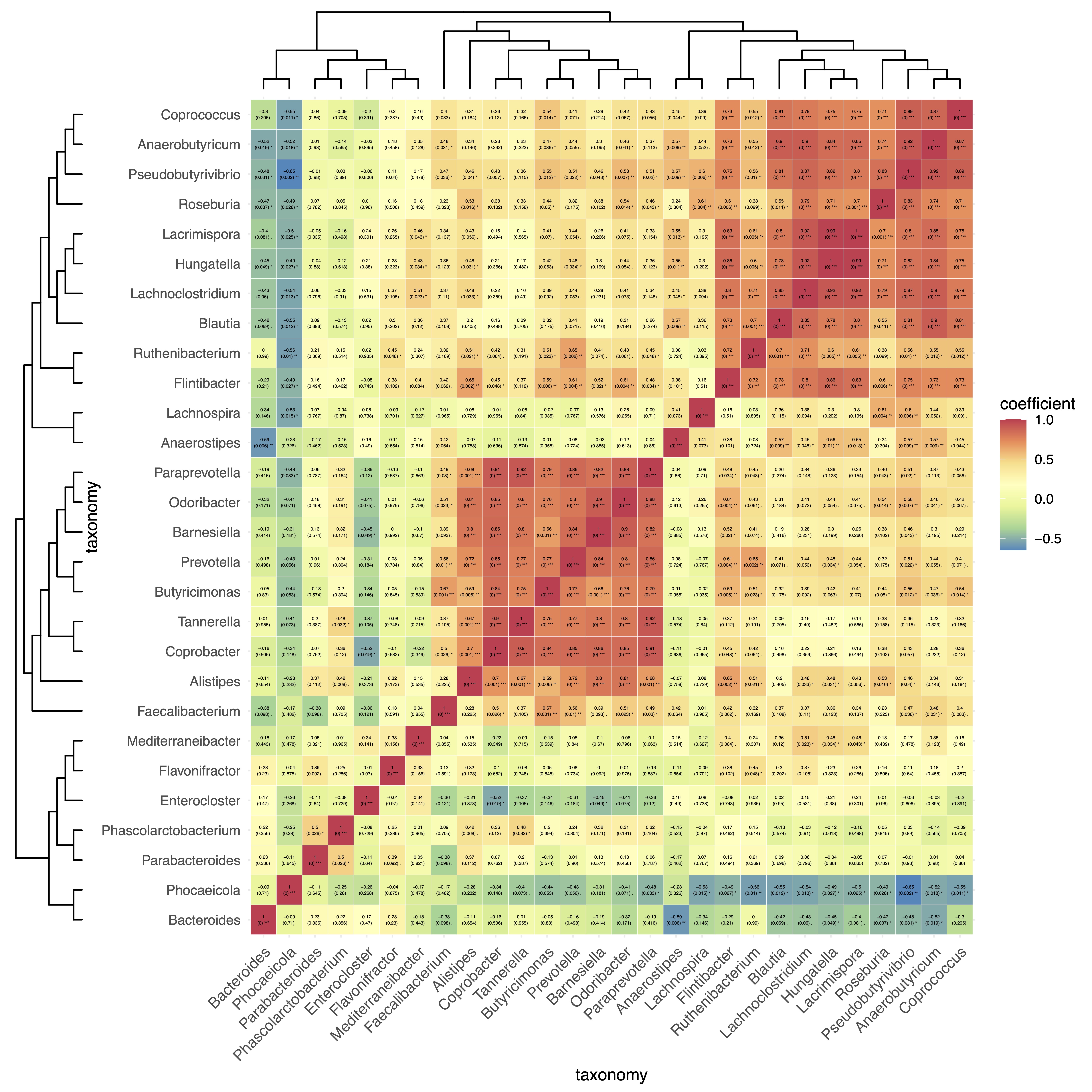
🏷️示例2:分析微生物之间的相互关系,即只选择一个组学构建自相关分析。
①这里使用
NULL可以构建只有一个组学的EMP对象。②当
EMP对象中仅包含一个组学时,将执行自相关计算。③如需绘制菌群的共发生网络图,可以先使用参数
action='get'获得相关性邻接矩阵,再导入到专业的网络分析工具(例如:Cytoscape 、MENA和Gephi等工具)进一步分析。
(micro_data + NULL) |>
EMP_cor_analysis() |>
EMP_heatmap_plot(label_size=1,palette='Spectral',clust_row=TRUE,clust_col=TRUE)
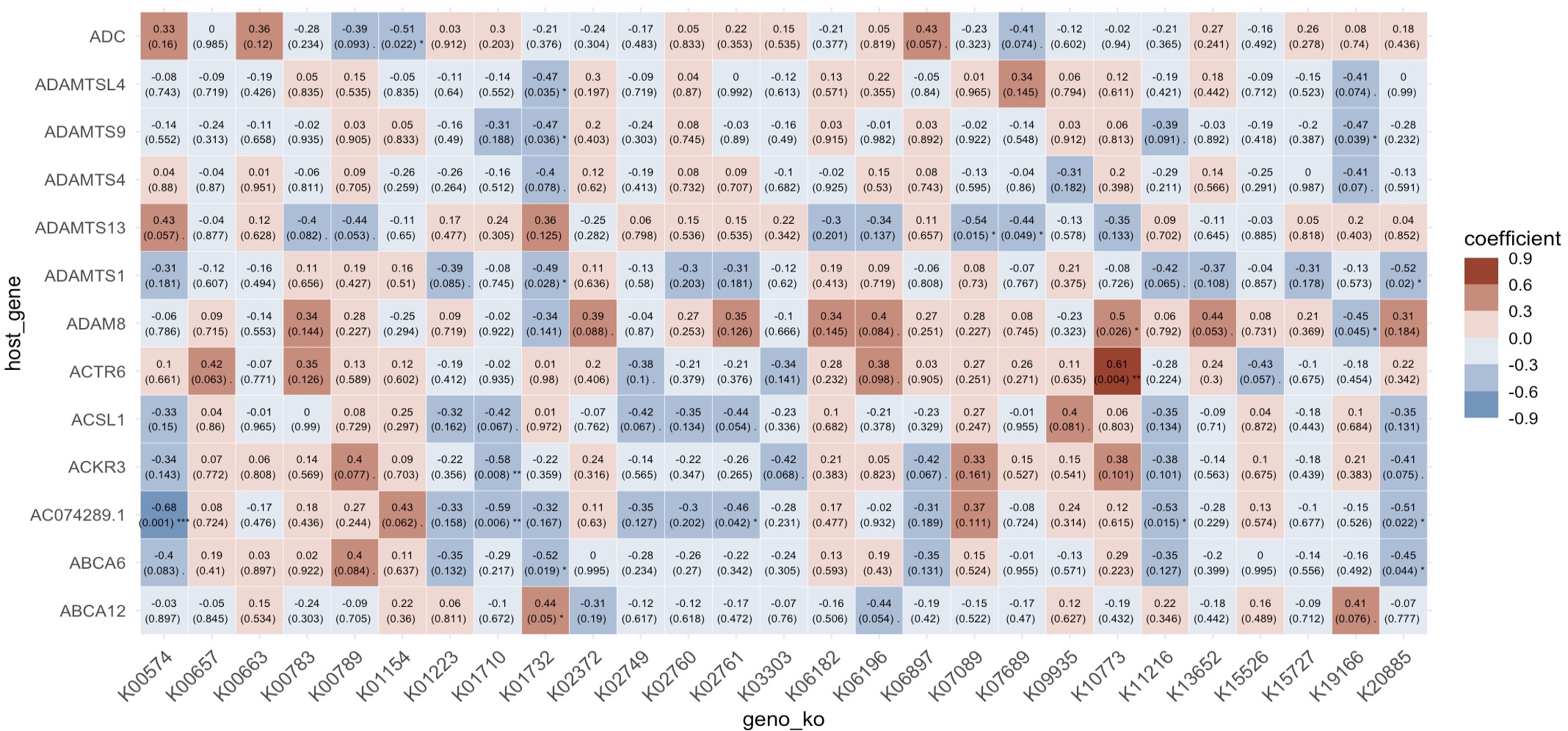
6.3.2 探索微生物差异功能基因与宿主差异基因表达的相关性
🏷️示例:微生物差异功能基因与宿主差异基因表达的相关性分析。
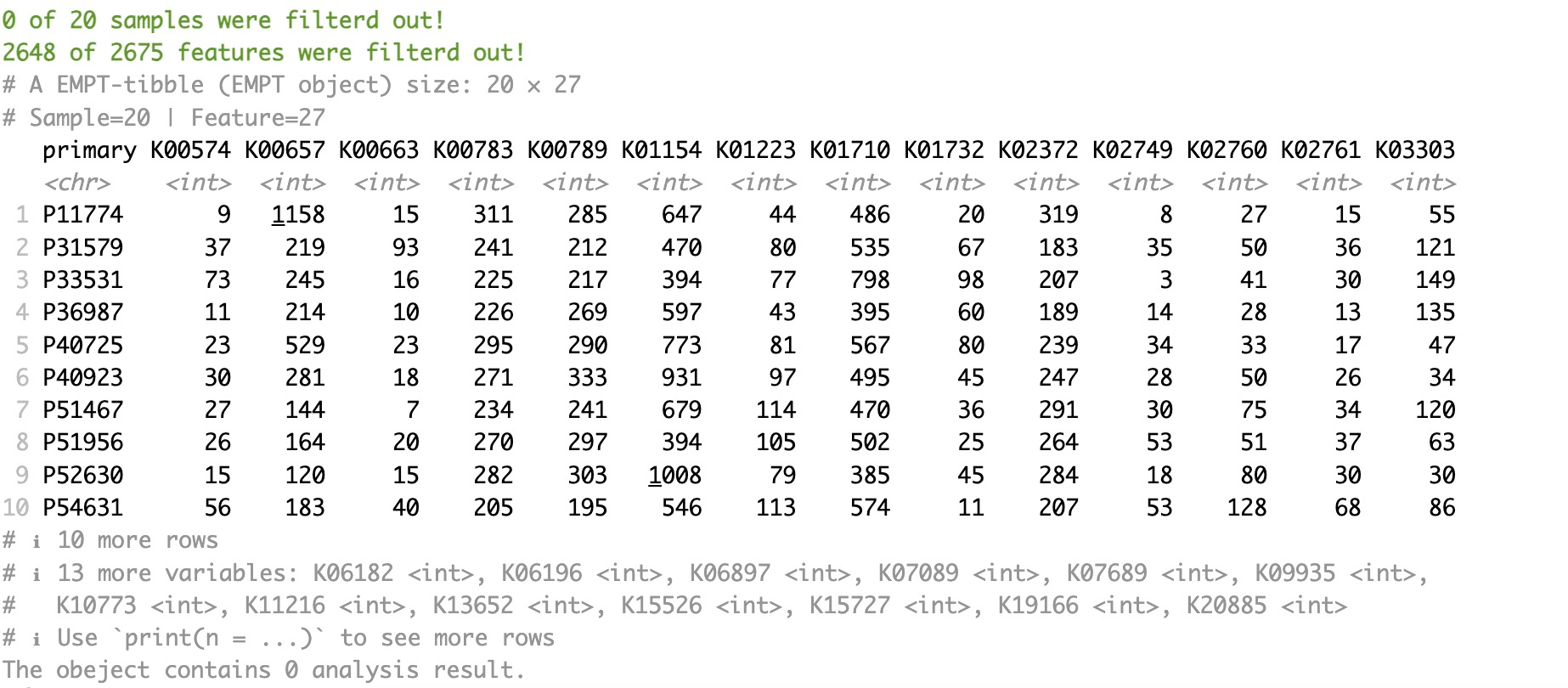
首先,从MAE对象中提取微生物差异功能的KO基因集,利用模块EMP_identify_assay过滤稀疏基因,利用模块EMP_diff_analysis 进行DESeq2差异性分析并考虑区域因素导致的批次问题,筛选出p值小于0.05的差异KO。
ko_data <- MAE |>
EMP_assay_extract('geno_ko') |>
EMP_identify_assay(method='edgeR') |>
EMP_diff_analysis(method='DESeq2',.formula = ~Region+Group) |>
EMP_filter(feature_condition = pvalue < 0.05)
ko_data
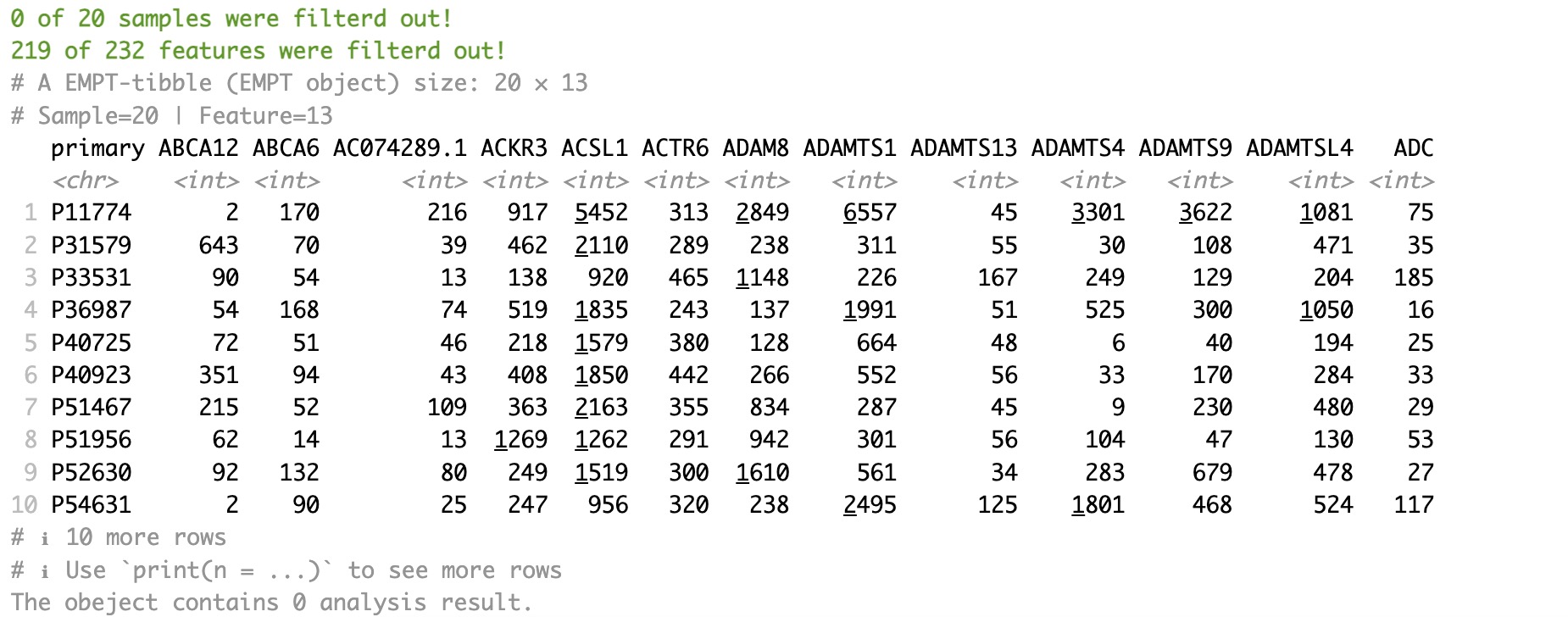
其次,从MAE对象中提取宿主转录组基因集,利用模块EMP_identify_assay过滤稀疏基因,利用模块EMP_diff_analysis 进行DESeq2差异性分析并考虑区域因素导致的批次问题,筛选出p值小于0.05的差异KO。
host_gene <- MAE |>
EMP_assay_extract('host_gene') |>
EMP_identify_assay(method='edgeR') |>
EMP_diff_analysis(method='DESeq2',.formula = ~Region+Group) |>
EMP_filter(feature_condition = pvalue < 0.05)
host_gene
最后,直接使用+符号将两个组学数据合并成EMP对象,使用模块EMP_cor_analysis和模块EMP_heatmap_plot完成相关性分析及可视化。
(ko_data + host_gene) |>
EMP_cor_analysis() |>
EMP_heatmap_plot()
6.3.3 探索多重相关性
模块EMP_cor_analysis能够计算多个组学项目数据之间的相互关系。我们可以分别计算各个组学项目数据之间的差异特征,使用+符号将组学合并在一起,并按照组合顺序进行相关性检验。模块EMP_sankey_plot可以根据多重相关性结果,绘制相关性桑基图。
①相关性桑基图中,红色为正相关,蓝色为负相关。
②相关性桑基图会评估每个节点之间的相互关系,孤立的节点将会被过滤。
③参数
pvalue和rvalue可以调节边的数量。
🏷️示例:探索微生物-功能基因-代谢产物-宿主基因-样本相关数据之间的多重相关性。
micro_data <- MAE |>
EMP_assay_extract('taxonomy') |>
EMP_identify_assay(method='default') |>
EMP_collapse(estimate_group = 'Genus',collapse_by = 'row') |>
EMP_decostand(method='relative')
ko_data <- MAE |>
EMP_assay_extract('geno_ko') |>
EMP_identify_assay(method='edgeR') |>
EMP_diff_analysis(method='DESeq2',.formula = ~Region+Group) |>
EMP_filter(feature_condition = pvalue < 0.05)
metabolite_data <- MAE |>
EMP_assay_extract(experiment = 'untarget_metabol') |>
EMP_collapse(estimate_group = 'MS2kegg',collapse_by='row',
na_string = c("NA", "null", "","-"),
method = 'mean',collapse_sep = '+') |>
EMP_decostand(method = 'relative') |>
EMP_dimension_analysis(method = 'pls',estimate_group = 'Group') |>
EMP_filter(feature_condition = VIP >2)
host_gene <- MAE |>
EMP_assay_extract('host_gene') |>
EMP_identify_assay(method='edgeR') |>
EMP_diff_analysis(method='DESeq2',.formula = ~Region+Group) |>
EMP_filter(feature_condition = pvalue < 0.05)
meta_data<- MAE |>
EMP_assay_extract('taxonomy') |>
EMP_coldata_extract(action = 'add',
coldata_to_assay = c('SAS','SDS','HAMA','HAMD','PHQ9','GAD7'))
(micro_data + ko_data + metabolite_data + host_gene + meta_data) |>
EMP_cor_analysis() |>
EMP_sankey_plot()
